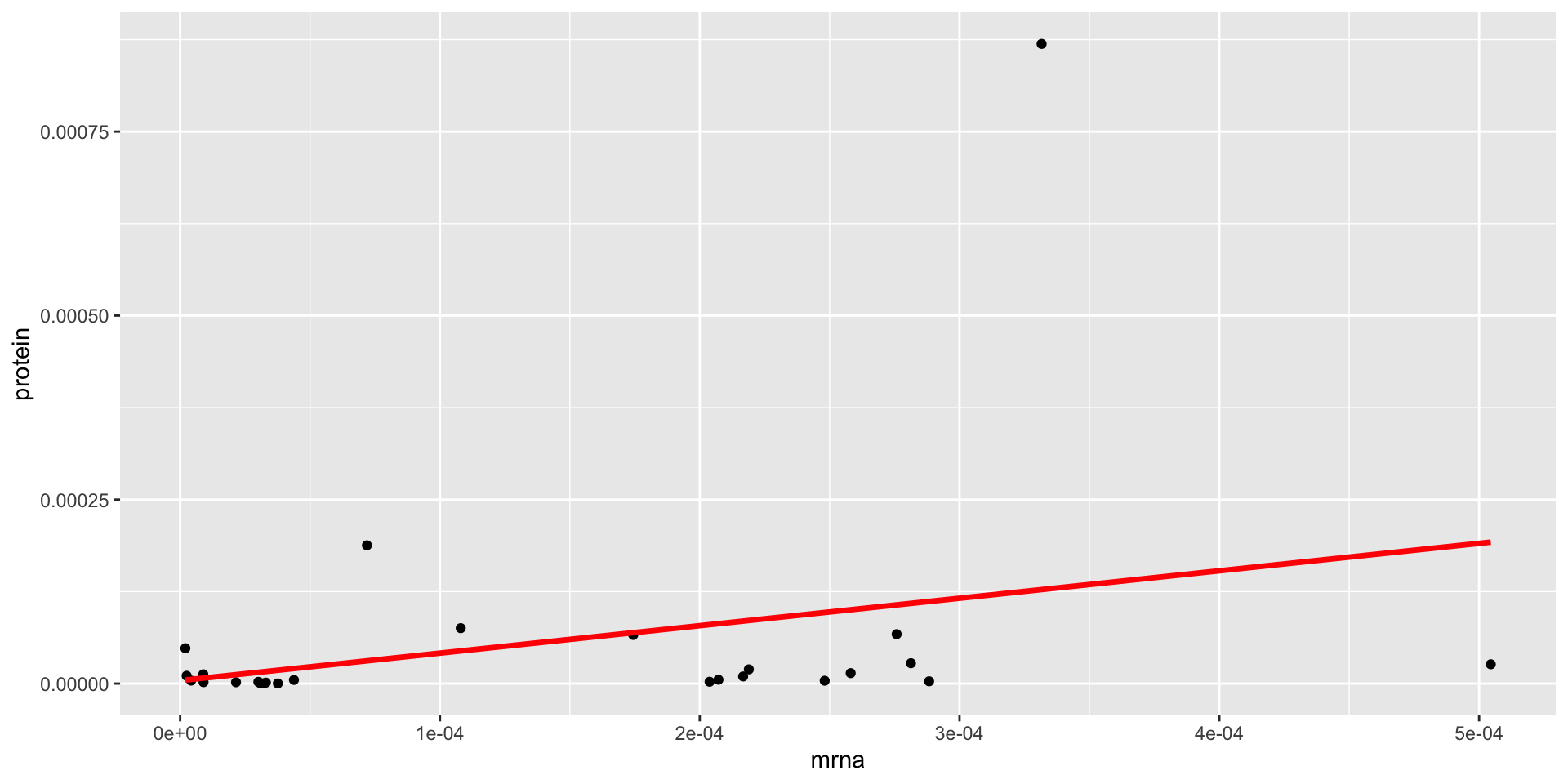
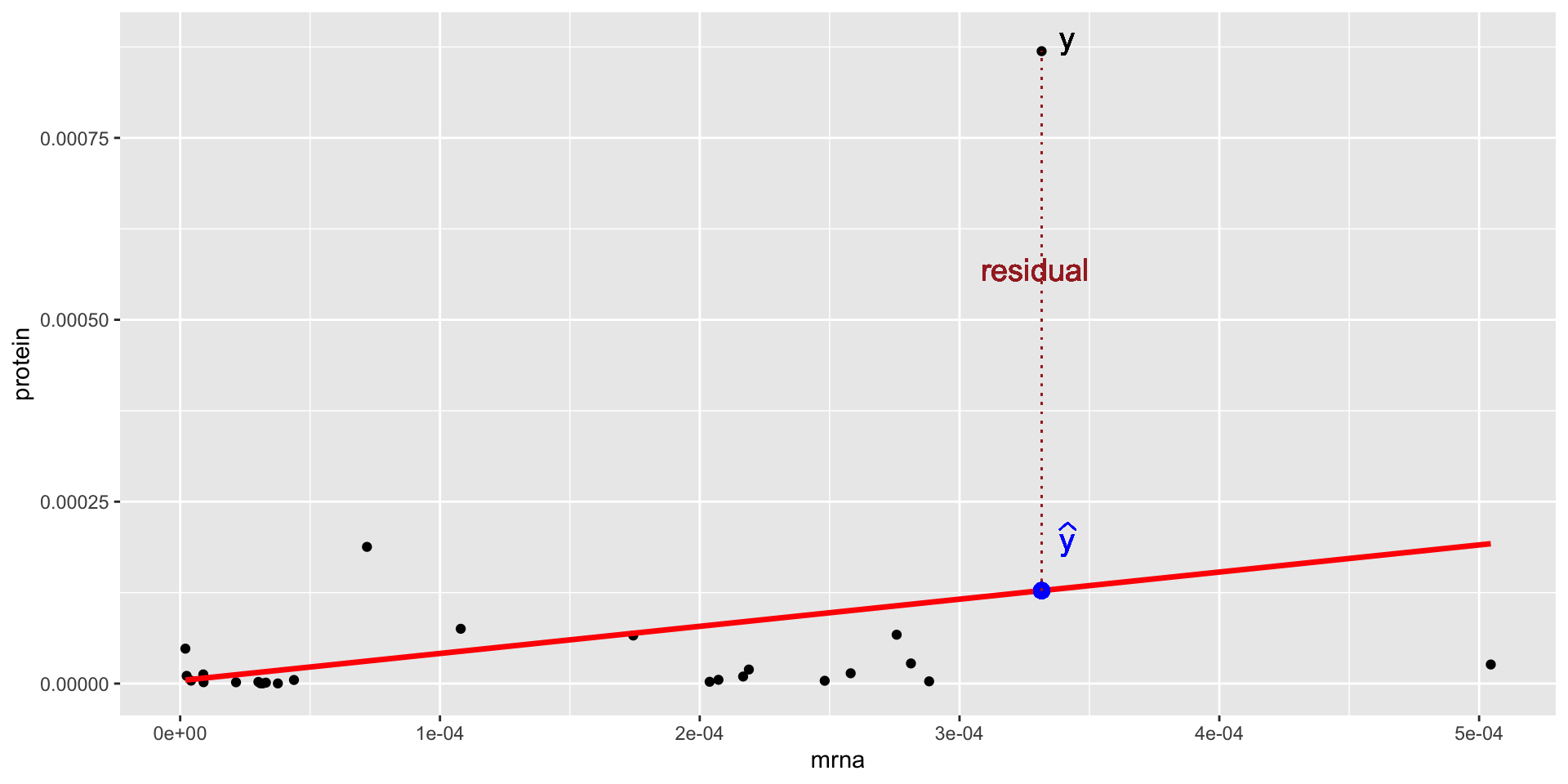
If our model provides a good fit, we expect the TSS (residuals from the null model, in red) to be much larger than the RSS (residuals from the fitted model, which we minimized by LS, in blue)!!
Picture from Wikipedia
Using the decomposition above and dividing by TSS:
For the majority of the genes, the SLR does not fit the data well
Is the \(R^2\) useful??
Scroll down to see full content
When we use LS, we get an \(R^2=0.56\), is this value large??
The \(R^2\) can be used to compare the size of the residuals of the fitted model with those of the null
The \(R^2\) can’t be used to test any hypothesis to answer this question since its distribution is unknown
The \(R^2\) is computed based on in-sample observations
The \(R^2\) does not provide a sense of how good is our model in predicting out-of-sample cases (aka test set)!!
Note that \(R^2\) computed as \(R^2 = 1 - \frac{\text{RSS}}{\text{TSS}}\) ranges between 0 and 1 if the LR model has an intercept and is estimated by LS!!
The \(R^2\) increases as new variables are added to the model, regardless of their relevance!! Thus, it can’t be used to compare nested models
Model Evaluation via Adjusted \(R^2\)
Scroll down to see full content
The \(R^2\) increases as more input variables are added to the model since the \(\text{RSS} = \sum_{i = 1}^n(y_i - \hat{y}_i)^2\) decreases as more input variables are included in the model.
To overcome this issue with \(R^2\), we can obtain an adjusted \(R^2\) as follows:
\(p\) is the number of covariates in the model (excluding the intercept)
\(n\) is our sample size to estimate the model
This adjusted coefficient of determination penalizes \(\text{RSS}\) with \(n - p - 1\). Hence, even if the \(\text{RSS}\) decreases, we divide it by \(n - p -1\) to compensate for the model’s size.
Other evaluation metrics
Scroll down to see full content
Other related metrics used to evaluate LR which measure how far \(Y\) is from \(\hat{Y}\) are:
Residuals Standard Error: RSE = \(\sqrt{\frac{1}{n-p - 1} \text{RSS}}\)
called sigma in glance() output
The RSE estimates the standard deviation of the error term \(\varepsilon\) (the RSS is divided by the appropriate degrees of freedom to give a “good” estimate of \(\sigma = \sqrt{Var(\varepsilon)}\))
The RSE is a measure based on training data to evaluate the fit of the model (for inference) and needed to estimate the standard errors of \(\hat{\beta}_j\) in classical theory!
The RSE gives an idea of the size of the irreducible error, very similar to the RSS, small is good
Mean Squared Error: MSE = \(\frac{1}{n}\sum_{i=1}^n(y_i - \hat{y}_i)^2\)
Training MSE: if we use the original sample \(y_1, \ldots, y_n\) and their predicted values (formula above). It can be easily obtained from .resid column in augment() output
Testing MSE: can be computed on new data \(y_{\text{new}}\) and their predicted values (formula above) to evaluate out-of-sample prediction performance
iClicker questions
Question 1: model.1
Which of the following codes would you use to estimate the model below: